This question has been answered in the comments by @thelatemail, but I've added some extra details for clarity.
One approach (as suggested by @thelatemail) is to subset the matrix to only include the gene you're interested in, e.g.
matrix_1 <- structure(c(0.3, 0, 0.4, 0.4, 0.1, 0.6, 0.8, 0.8, 0.9,
1.3, 0.9, 0.1, 3.3, 0.9, 0.1),
.Dim = c(3L, 5L),
.Dimnames = list(c("gene_1", "gene_2", "gene_3"),
c("X1", "X2", "X3", "X4", "X5")))
matrix_1
#> X1 X2 X3 X4 X5
#> gene_1 0.3 0.4 0.8 1.3 3.3
#> gene_2 0.0 0.1 0.8 0.9 0.9
#> gene_3 0.4 0.6 0.9 0.1 0.1
df_1 <- structure(list(Sample = 1:5,
PEDIS = c("PEDIS1", "PEDIS1", "PEDIS2",
"PEDIS2", "PEDIS2")),
class = c("tbl_df", "tbl", "data.frame"),
row.names = c(NA, -5L))
df_1
#> # A tibble: 5 × 2
#> Sample PEDIS
#> <int> <chr>
#> 1 1 PEDIS1
#> 2 2 PEDIS1
#> 3 3 PEDIS2
#> 4 4 PEDIS2
#> 5 5 PEDIS2
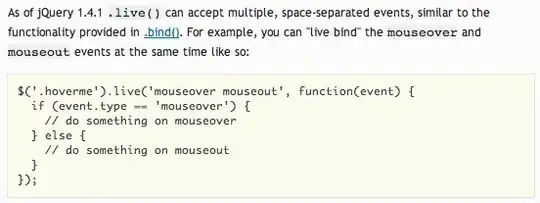
# Boxplot
boxplot(matrix_1[2,] ~ df_1$PEDIS)
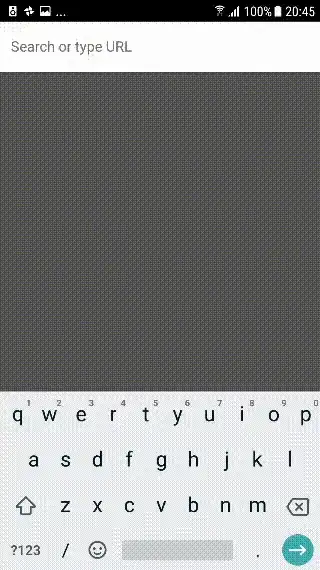
# Or, using the gene name, rather than the row number
boxplot(matrix_1["gene_2",] ~ df_1$PEDIS)
Created on 2021-09-09 by the reprex package (v2.0.1)
--
Another approach is to combine and transpose your data into the 'tidy' format (each row is a sample, each column is a variable), e.g.
df_2 <- as.data.frame(t(matrix_1))
df_2$group <- df_1$PEDIS
df_2
#> gene_1 gene_2 gene_3 group
#> X1 0.3 0.0 0.4 PEDIS1
#> X2 0.4 0.1 0.6 PEDIS1
#> X3 0.8 0.8 0.9 PEDIS2
#> X4 1.3 0.9 0.1 PEDIS2
#> X5 3.3 0.9 0.1 PEDIS2
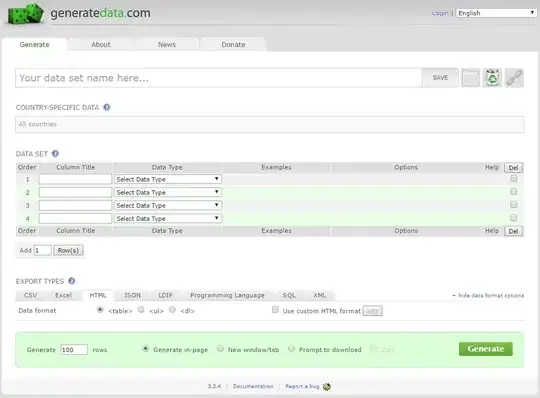
boxplot(gene_2 ~ group, data = df_2)
The tidy format facilitates the use of the ggplot2 package (for details on why this is a good thing, see: https://r-graphics.org/) which enables you to add 'features' to your plot, e.g. the individual logCPM values (dots) for each sample:
library(tidyverse)
df_2 %>%
pivot_longer(cols = -group, names_to = "genes") %>%
filter(genes == "gene_2") %>%
ggplot(aes(x = group, y = value)) +
geom_boxplot(outlier.shape = NA) +
geom_jitter(aes(color = genes), width = 0.2) +
theme_minimal()

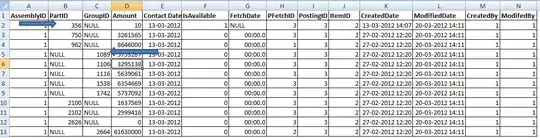
You can also make more complicated figures, for example, comparing logCPM values for two genes 'side-by-side':
df_2 %>%
pivot_longer(cols = -group, names_to = "genes") %>%
filter(genes %in% c("gene_1", "gene_2")) %>%
ggplot(aes(x = group, y = value)) +
geom_boxplot(outlier.shape = NA) +
geom_jitter(aes(color = genes), width = 0.2) +
theme_minimal() +
facet_wrap(~ genes)
Created on 2021-09-09 by the reprex package (v2.0.1)
